MicroRNA-33 inhibition ameliorates muscular dystrophy by enhancing skeletal muscle regeneration
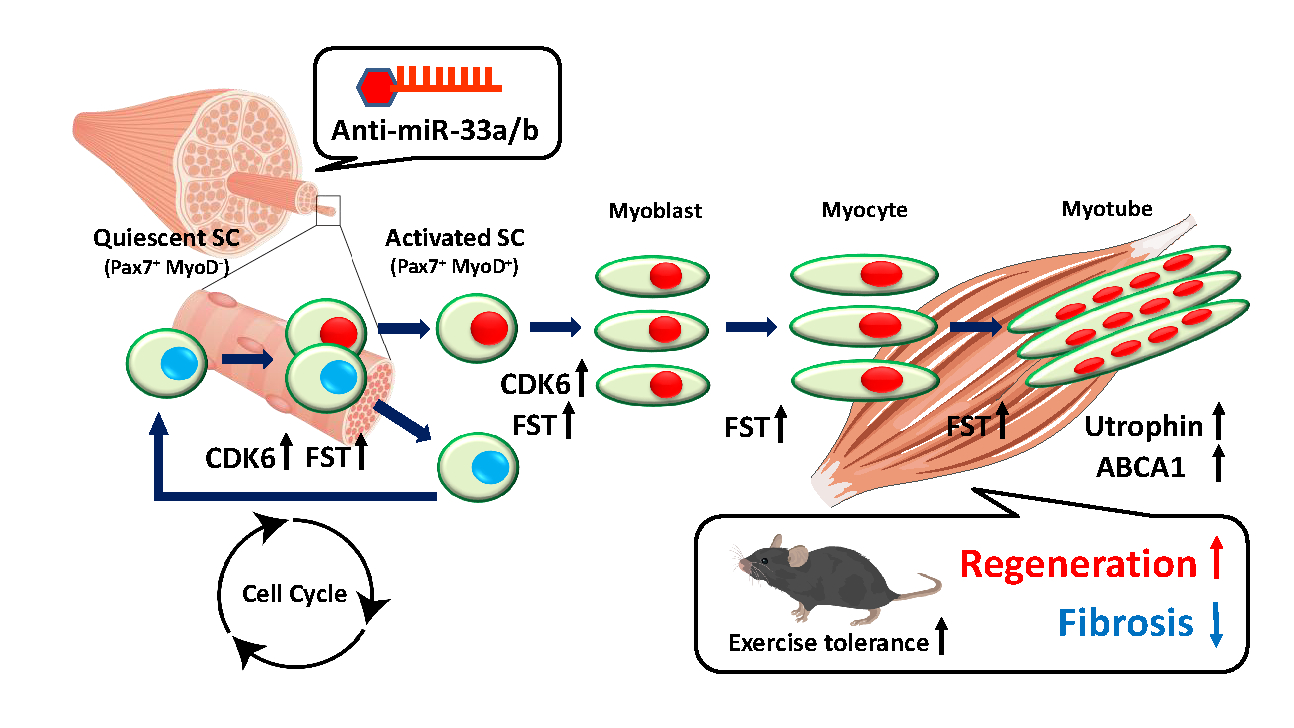
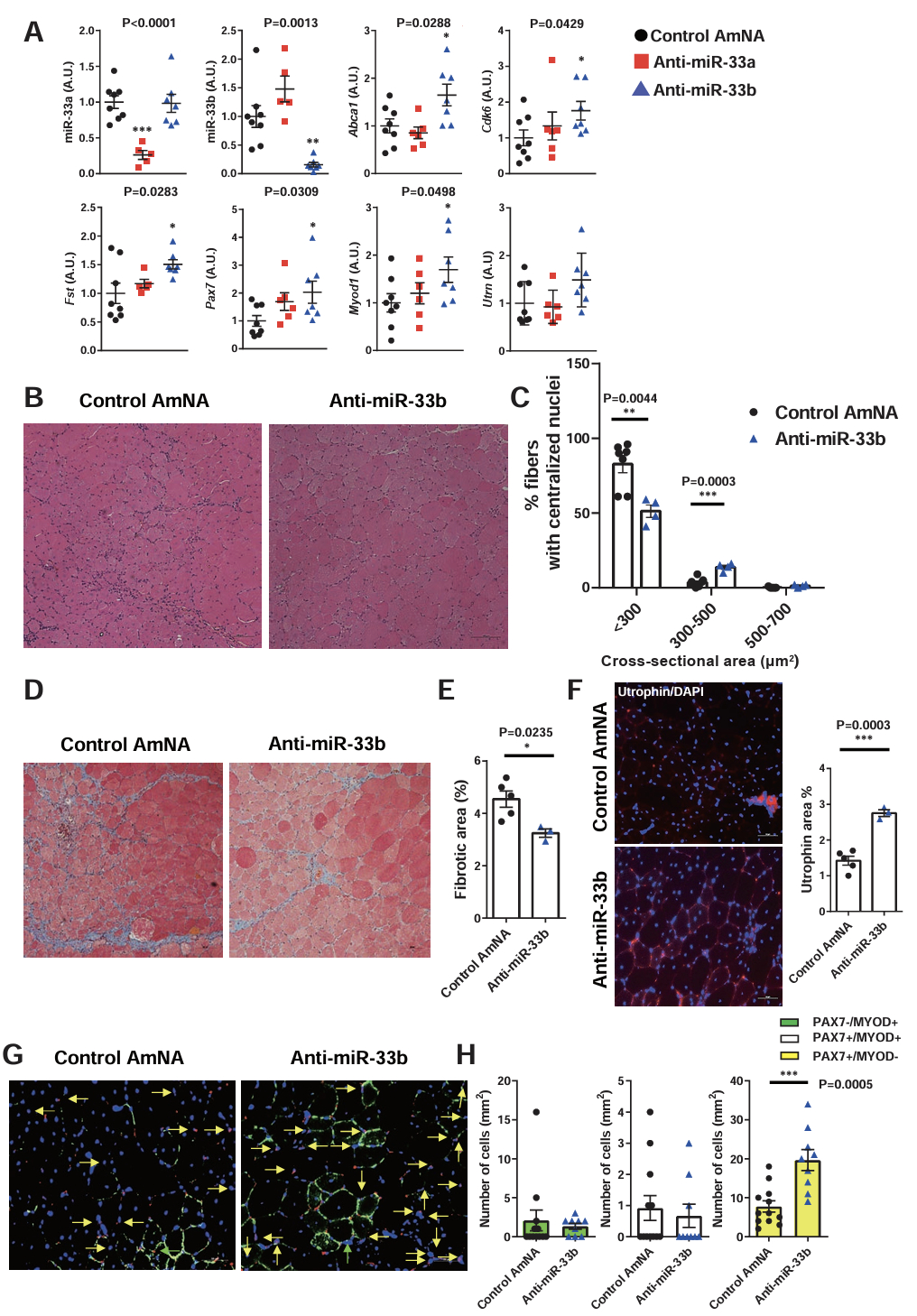
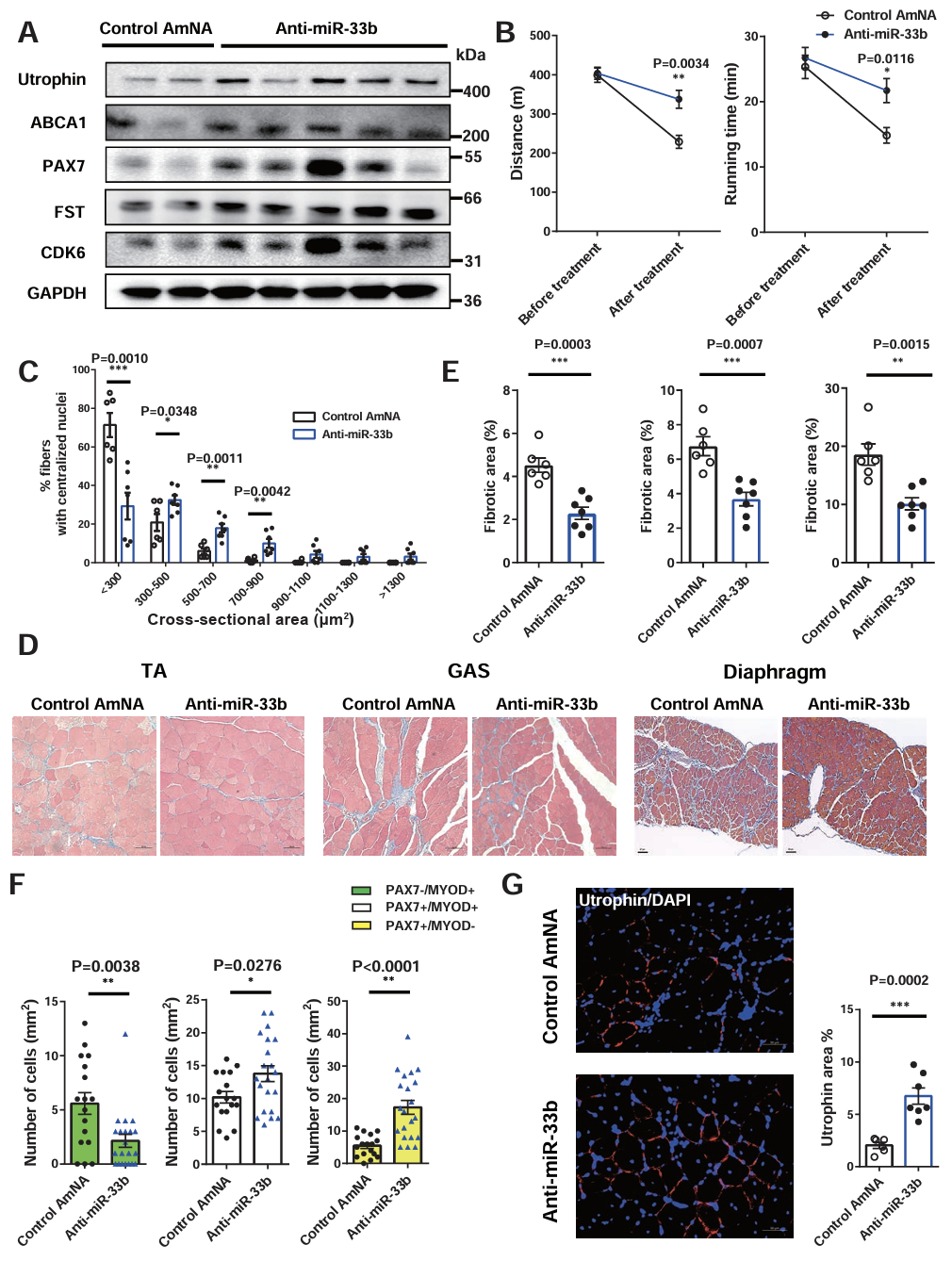
Abstract Body: Background: Muscular dystrophy involves genetic diseases causing progressive skeletal muscle weakness. Duchenne muscular dystrophy (DMD) is a severe form caused by dystrophin deficiency, leading to muscle damage, inflammation, and fibrosis that inhibit regeneration. MicroRNAs (miRNAs) modulate muscle repair. miR-33a/b's specific role in muscle regeneration remains unclear. Purpose: This study elucidates the precise function of miR-33a/b in skeletal muscle regeneration and evaluates whether pharmacological inhibition of miR-33 represents a viable therapeutic strategy for DMD. Methods: Researchers analyzed mouse models to dissect miR-33 function. miR-33a-knockout (KO) mice were evaluated using a cardiotoxin (CTX)-induced injury model and crossed with mdx mice (a DMD model). Humanized miR-33b knock-in (KI) mdx mice were generated to mimic human physiology, expressing both miR-33a and miR-33b. The team assessed satellite cell (SC) proliferation, histology, and target genes (Cdk6, Fst, Abca1). AAV9-mediated shRNA was used for rescue experiments. Anti-miRNA oligonucleotides (AMOs) against miR-33a/b were tested in mdx mice and human DMD iPS cell-derived myotubes to gauge therapeutic efficacy. Results: In CTX-injury and mdx models, miR-33a-KO mice showed accelerated muscle regeneration, reduced fibrosis, increased SC proliferation, elevated utrophin, and improved endurance. Conversely, miR-33b-KI mdx mice exhibited exacerbated dystrophic phenotypes with weaker regenerative capacity. Mechanistically, miR-33a/b negatively regulate SC proliferation and muscle regeneration by targeting the 3'-UTR of Cdk6, Fst, and Abca1. AAV9-mediated knockdown of these genes reversed the beneficial phenotypes in miR-33a-KO mice. Importantly, systemic AMO administration suppressed miR-33 activity, upregulated target genes, and remarkably improved muscle pathology and running capacity in mdx mice. Also, anti-miR-33b treatment in human DMD iPS cell myotubes upregulated CDK6, FST, ABCA1, and utrophin, mirroring the in vivo findings. Conclusions: miR-33a/b are critical negative regulators of muscle regeneration. Inhibiting miR-33 profoundly enhances SC expansion and attenuates fibrosis by targeting Cdk6, Fst, and Abca1. AMO-mediated miR-33 inhibition is therefore a promising therapeutic approach for managing DMD.
Kojima, Hidenori
(
Kyoto University
, Kyoto , Japan )
Sowa, Naoya
(
kyoto univ. cardiovascular med.
, Kyoto Japan , Japan )
Horie, Takahiro
(
KYOTO UNIVERSITY HOSPITAL
, Kyoto , Japan )